Top 7 ab initio calculations in 2023
Below are the best information and knowledge on the subject ab initio calculations compiled and compiled by our own team dvn:
Mục Lục
1. Ab_initio_quantum_chemistry_methods
Author: www.sciencedirect.com
Date Submitted: 06/24/2021 08:39 AM
Average star voting: 5 ⭐ ( 18562 reviews)
Summary: Ab initio quantum chemistry methods Ab initio quantum chemistry methods are computational chemistry methods based on quantum chemistry.[1] The term ab initio
Match with the search results: Ab initio calculations are computations of electronic orbitals with no other hypotheses than Coulomb interactions between all electrons and nuclei with electrons obeying Fermi statistics with the Pauli exclusion principle….. read more

2. Ab initio Calculations
Author: en.wikipedia.org
Date Submitted: 06/03/2020 01:04 AM
Average star voting: 3 ⭐ ( 71090 reviews)
Summary: Ab initio calculations rest on solving the Schrödinger equation; the nature of the necessary approximations determines the level of the calculation. In the simplest approach, the Hartree-Fock method, the total molecular wavefunction Ψ is approximated as a…
Match with the search results: Ab initio quantum chemistry methods attempt to solve the electronic Schrödinger equation given the positions of the nuclei and the number of electrons in order ……. read more

3. Ab initio calculation of real solids via neural network ansatz | Nature Communications
Author: www.oxfordreference.com
Date Submitted: 03/03/2019 09:50 AM
Average star voting: 4 ⭐ ( 10753 reviews)
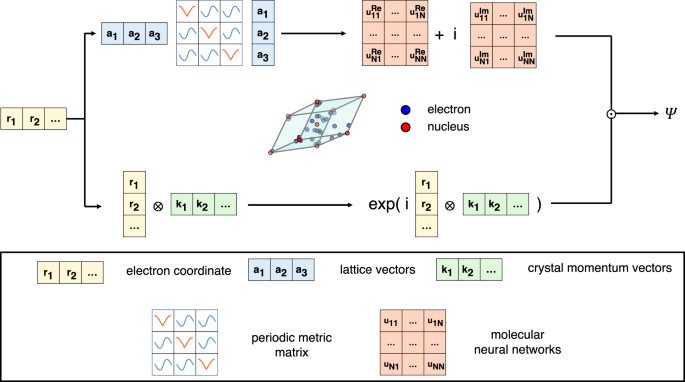
Summary: Neural networks have been applied to tackle many-body electron correlations for small molecules and physical models in recent years. Here we propose an architecture that extends molecular neural networks with the inclusion of periodic boundary conditions to enable ab initio calculation of real solids. The accuracy of our approach is demonstrated in four different types of systems, namely the one-dimensional periodic hydrogen chain, the two-dimensional graphene, the three-dimensional lithium hydride crystal, and the homogeneous electron gas, where the obtained results, e.g. total energies, dissociation curves, and cohesive energies, reach a competitive level with many traditional ab initio methods. Moreover, electron densities of typical systems are also calculated to provide physical intuition of various solids. Our method of extending a molecular neural network to periodic systems can be easily integrated into other neural network structures, highlighting a promising future of ab initio solution of more complex solid systems using neural network ansatz, and more generally endorsing the application of machine learning in materials simulation and condensed matter physics. Solving the many-body electronic structure of real solids is a grand challenge in condensed matter physics and materials science. Here authors present a machine learning ab initio architecture for real solids, which combines molecular neural network wavefunction ansatz and periodic features, providing accurate solutions for a range of solids.
Match with the search results: Ab initio quantum chemistry methods are computational chemistry methods based on quantum chemistry. The term ab initio was first used in quantum chemistry by Robert Parr and coworkers, including David Craig in a semiempirical study on the excited……. read more
4. Ab Initio Calculations
Author: www.chemeurope.com
Date Submitted: 03/24/2019 04:47 PM
Average star voting: 4 ⭐ ( 12149 reviews)
Summary: Abstract. Various difficulties of classical physics, including inadequate description of atoms and molecules, led to new ways of visualizing physical realities,
Match with the search results: Periodic boundary conditions in ab initio calculations…. read more

5. Can DFT be considered an ab initio method?
Author: www.pnas.org
Date Submitted: 03/06/2021 10:26 PM
Average star voting: 3 ⭐ ( 71925 reviews)
Summary:
Match with the search results: A method of calculating atomic and molecular structure directly from the first principles of quantum mechanics, without using quantities derived from ……. read more

6. Phys. Rev. A 104, 052810 (2021) – Ab initio calculations of the spectrum of lawrencium
Author: link.springer.com
Date Submitted: 04/27/2022 04:44 PM
Average star voting: 5 ⭐ ( 30066 reviews)
Summary: We present high-accuracy relativistic investigations of the spectrum of Lr, element 103, prompted by the planned optical spectroscopy experiments on this rare and short-lived atom. Reliable predictions of the transition lines are important for the planning and success of these challenging measurements. The relativistic coupled cluster approach was used to calculate the energies of lowest excited states, while the combination of configuration-interaction method with the many-body perturbation theory was employed to address the higher-lying states and to obtain the transition strengths and the lifetimes of the levels of experimental interest. We performed similar calculations for Lu, the lighter homologue of Lr, where experimental data are available. For the lighter element, both the calculated energies and the Einstein coefficients are in excellent agreement with the previously measured values, confirming the accuracy of the performed calculations and the reliability of our predictions for Lr.
Match with the search results: The term ab initio indicates that the calculation is from first principles and that no empirical data is used. Robert Parr claims in an interview that the term ……. read more

7. Phys. Rev. C 105, 014302 (2022) – Converged ab initio calculations of heavy nuclei
Author: www.youtube.com
Date Submitted: 06/28/2022 12:32 AM
Average star voting: 3 ⭐ ( 46935 reviews)
Summary: We propose a novel storage scheme for three-nucleon (3N) interaction matrix elements relevant for the normal-ordered two-body approximation used extensively in ab initio calculations of atomic nuclei. This scheme reduces the required memory by approximately two orders of magnitude, which allows the generation of 3N interaction matrix elements with the standard truncation of ${E}_{3\mathrm{max}}=28$, well beyond the previous limit of 18. We demonstrate that this is sufficient to obtain the ground-state energy of $^{132}\mathrm{Sn}$ converged to within a few MeV with respect to the ${E}_{3\mathrm{max}}$ truncation. In addition, we study the asymptotic convergence behavior and perform extrapolations to the un-truncated limit. Finally, we investigate the impact of truncations made when evolving free-space 3N interactions with the similarity renormalization group. We find that the contribution of blocks with angular momentum ${J}_{\mathrm{rel}}>9/2$ to the ground-state energy is dominated by a basis-truncation artifact, which vanishes in the large-space limit, so these computationally expensive components can be neglected. For the two sets of nuclear interactions employed in this work, the resulting binding energy of $^{132}\mathrm{Sn}$ agrees with the experimental value within theoretical uncertainties. This work enables converged ab initio calculations of heavy nuclei.
Match with the search results: Ab initio calculations provide a wealth of detail that is not available from experiment and a degree of confidence in the results that is not ……. read more